7 Min Read
7 Min Read
Thalassemia is a hereditary blood disorder that affects the body’s ability to produce haemoglobin, the oxygen-carrying component of red blood cells. When haemoglobin production is disrupted, the body cannot deliver enough oxygen to tissues, resulting in anaemia that can range from mild to life-threatening.
At its core, haemoglobin is made up of four protein chains, two alpha (α) and two beta (β). Thalassemia occurs when mutations affect the genes responsible for producing these chains. Since these mutations are inherited from parents, the condition is present from birth and varies significantly in severity depending on how many genes are affected.
In countries like India, where thalassemia prevalence is relatively high, awareness and early diagnosis play a crucial role in managing the condition effectively.
Synopsis
What Causes Thalassemia?
Thalassemia is not caused by lifestyle or environmental factors; it is purely genetic. The disorder arises when there are mutations or deletions in the genes responsible for haemoglobin production, leading to either reduced or absent production of alpha or beta globin chains.
There are two primary types of thalassemia, classified based on which globin chain is affected:
Alpha vs Beta Thalassemia: Understanding the Difference
|
Feature |
Alpha Thalassemia |
Beta Thalassemia |
|
Affected Genes |
HBA1 and HBA2 |
HBB gene |
|
Chromosome Location |
Chromosome 16 |
Chromosome 11 |
|
Severity Basis |
Number of genes affected (1–4) |
Number of faulty genes (1 or 2) |
|
Common Outcome |
Mild to fatal (in severe cases) |
Mild anaemia to severe transfusion-dependent condition |
In alpha thalassemia, the severity increases with the number of affected genes. A person with one defective gene may not even know they carry the condition, while all four affected genes can lead to a fatal condition before or shortly after birth.
In contrast, beta thalassemia requires two defective genes, one inherited from each parent, for the severe form to manifest. Individuals with only one faulty gene are typically carriers (thalassemia minor) and may experience only mild anaemia.
Who Is at Risk?
Thalassemia is more common among individuals from certain ethnic backgrounds. Populations from South Asia, the Mediterranean region, the Middle East, and Africa show higher prevalence. Interestingly, this distribution is historically linked to protection against malaria, which allowed these gene mutations to persist over generations.
Symptoms of Thalassemia: From Silent to Severe
The symptoms of thalassemia are not uniform; they depend heavily on the type and severity of the condition. While some individuals may live without ever realizing they have it, others may experience serious complications early in life.
Symptom Progression by Severity
|
Severity Level |
Clinical Presentation |
|
Mild |
Often asymptomatic, occasional fatigue or weakness |
|
Moderate |
Enlarged spleen/liver, delayed growth, bone deformities |
|
Severe |
Life-threatening anaemia usually appears within the first 2 years |
In mild thalassemia, individuals may experience only slight fatigue and often lead completely normal lives without requiring treatment.
However, as the condition progresses to moderate levels, symptoms become more visible. Children may experience delayed physical development, enlargement of organs like the spleen or liver, and even structural changes in bones, particularly in the face.
In severe thalassemia (thalassemia major), symptoms are pronounced and often appear within the first two years of life. These include:
-
Extreme fatigue and weakness
-
Pale or yellowish skin (due to anaemia or jaundice)
-
Poor growth and developmental delays
-
Enlarged spleen causing abdominal swelling
-
Dark urine due to the rapid breakdown of red blood cells
-
Frequent infections due to compromised immunity
Without timely intervention, severe thalassemia can become life-threatening.
How Is Thalassemia Diagnosed?
Early and accurate diagnosis is critical in managing thalassemia effectively. The diagnostic process involves a combination of clinical evaluation, laboratory testing, and genetic analysis.
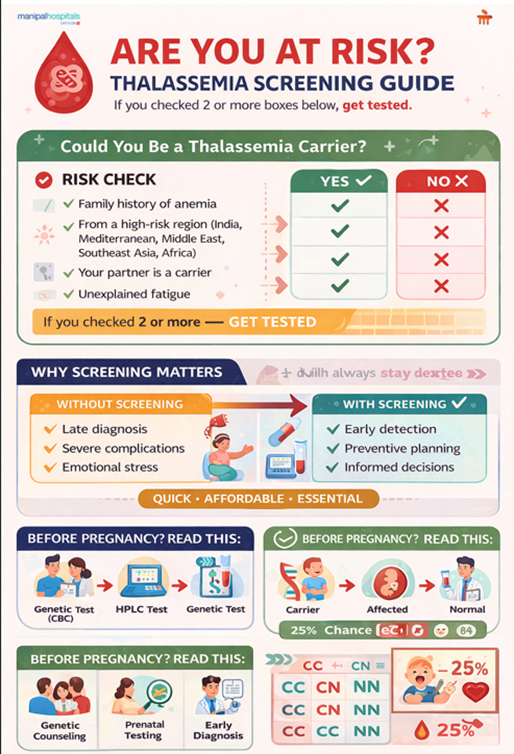
Step-by-Step Diagnostic Approach
Doctors often begin with a clinical suspicion, especially when a child presents with anaemia, pale skin, or delayed growth. A family history of blood disorders and belonging to high-risk ethnic groups further strengthens this suspicion.
This is followed by laboratory investigations:
|
Test |
Role in Diagnosis |
Key Findings |
|
Complete Blood Count (CBC) |
Initial screening |
Low haemoglobin, small RBCs (microcytosis) |
|
Peripheral Blood Smear |
Morphological analysis |
Target cells, teardrop-shaped cells |
|
Iron Studies |
Rule out iron defiency |
Normal or high iron levels |
|
HPLC / Hemoglobin Electrophoresis |
Confirmatory test |
Detects abnormal haemoglobin types |
While these tests help identify the presence of thalassemia, genetic testing provides definitive confirmation. DNA analysis identifies mutations in specific genes, HBA1/HBA2 for alpha thalassemia and HBB for beta thalassemia.
Importance of Prenatal Diagnosis
For couples at risk (especially if both are carriers), prenatal testing becomes essential.
|
Procedure |
Timing |
Purpose |
|
Chorionic Villus Sampling (CVS) |
10-12 weeks |
Early detection of genetic abnormalities |
|
Amniocentesis |
15-18 weeks |
Confirms fetal diagnosis |
Prenatal HPLC can detect the thalassemia carrier state in the mother. When the mother is known to be a carrier, the father is to be tested. If the father is also a carrier, then proceed with genetic tests for the fetus. In India, prenatal diagnosis is increasingly being adopted in high-prevalence states like Gujarat, Maharashtra, and West Bengal, helping families make informed decisions.
Treatment Options: Managing Thalassemia Effectively
The treatment of thalassemia depends on its severity. While mild cases may not require any intervention, moderate to severe cases need ongoing medical care under the supervision of an experienced specialist.
|
Treatment |
Purpose |
Applicability |
|
Regular Monitoring |
Track haemoglobin levels |
Mild cases |
|
Blood Transfusions |
Maintain adequate haemoglobin |
Moderate to severe cases |
|
Iron Chelation Therapy |
Remove excess iron from the body |
Patients receiving transfusions |
|
Folic Acid Supplements |
Support red blood cell production |
Most patients |
|
Splenectomy |
Reduce complications from an enlarged spleen |
Selected cases |
|
Stem Cell Transplant |
Potential cure |
Eligible patients with donors |
|
Gene Therapy (CASGEVY™, 2024) |
One-time advanced treatment |
Transfusion-dependent beta thalassemia |
Treatment Overview
Among these, blood transfusions are the cornerstone of treatment for severe thalassemia. However, repeated transfusions lead to iron overload, which can damage vital organs such as the heart and liver.
This makes iron chelation therapy equally important, as it helps remove excess iron and prevent long-term complications.
A stem cell transplant remains the only established cure, but it requires a compatible donor and carries certain risks. More recently, gene therapy (CASGEVY™, approved in 2024) has emerged as a promising one-time treatment for specific patients, marking a major breakthrough in thalassemia care.
Complications to Watch Out For
If not managed properly, thalassemia can lead to serious health complications, primarily due to iron overload.
Common Long-Term Complications
-
Heart disease
-
Liver damage
-
Hormonal imbalances
-
Bone deformities
-
Growth delays
Among these, iron overload remains the most critical risk, especially in patients undergoing regular transfusions.
Can Thalassemia Be Prevented?
Since thalassemia is a genetic condition, it cannot be prevented in the traditional sense. However, its occurrence can be significantly reduced through awareness and screening.
Preventive Strategies That Matter
-
Genetic counselling before marriage or pregnancy
-
Carrier screening for at-risk individuals
-
Prenatal testing for early detection
These steps are particularly important in high-risk populations, where early intervention can prevent severe cases.
Prognosis: Living with Thalassemia
The outlook for individuals with thalassemia has improved dramatically over the years.
|
Type |
Outlook |
|
Mild |
Normal life expectancy, minimal intervention |
|
Moderate |
Managed with regular care |
|
Severe |
Lifelong treatment is required, but survival is improving |
Today, with advancements in diagnostics, transfusion protocols, iron chelation, and gene therapy, individuals with thalassemia can lead longer, healthier lives than ever before.
Ending Thoughts
At Varthur Road, patients with thalassemia receive comprehensive, patient-centric care backed by advanced diagnostics and multidisciplinary expertise. From early screening and accurate diagnosis using state-of-the-art technologies to personalised treatment plans that include transfusion support, iron chelation therapy, and access to evolving treatment options, the hospital ensures continuity of care at every stage, with guidance from experienced specialists. What truly sets Manipal Hospitals apart is its emphasis on preventive screening, genetic counselling, and family education, helping not just patients, but entire families make informed healthcare decisions. With a commitment to clinical excellence and compassionate care, Manipal Hospitals, Varthur Road, stands as a trusted partner in managing thalassemia and improving long-term patient outcomes.
FAQ's
Thalassemia is a genetic blood disorder that reduces the body’s ability to produce haemoglobin, leading to anaemia. This affects oxygen delivery to tissues, causing symptoms like fatigue, weakness, and in severe cases, organ complications.
Symptoms vary based on severity. Mild cases may show few to no symptoms, while moderate to severe cases can include fatigue, pale or yellowish skin, delayed growth, enlarged spleen, bone deformities, and frequent infections.
Diagnosis involves blood tests like a complete blood count (CBC), peripheral smear, iron studies, and confirmatory tests such as haemoglobin electrophoresis or HPLC. Genetic testing is used for definitive diagnosis, especially in prenatal cases.
Manipal Hospitals Varthur Road offers comprehensive, patient-centric care with advanced diagnostics, multidisciplinary expertise, and personalised treatment plans. The hospital also emphasises genetic counselling, preventive screening, and long-term management, ensuring better outcomes for both patients and families.
Share this article on: